崪奿嬝偵偍偗傞RyR1娭楢嬝幘姵偺敪徢婡峔偺夝柧偲憂栻
1宆儕傾僲僕儞庴梕懱乮RyR1乯偼崪奿嬝嬝彫朎懱偺Ca2+梀棧僠儍僱儖偱丄嫽暠廂弅楢娭偵廳梫側栶妱傪壥偨偟偰偄傑偡丅RyR1偼憤暘巕検偑2 MDa傪挻偊傞嫄戝側枌僞儞僷僋幙偱丄偦偺堚揱巕堎忢偼埆惈崅擬徢傗庬乆偺儈僆僷僠乕傪堷偒婲偙偟傑偡丅傢偨偟偨偪偼嵶朎撪Ca2+僀儊乕僕儞僌朄傪偼偠傔偲偟偨婡擻夝愅偵傛傝丄Ca2+摦懺堎忢偑嬝幘姵傪堷偒婲偙偡儊僇僯僘儉偍傛傃帯椕朄傪専摙偟偰偄傑偡丅
A. 崪奿嬝偵偍偗傞1宆儕傾僲僕儞庴梕懱乮RyR1乯偵傛傞Ca2+桿敪惈Ca2+梀棧乮CICR乯偺栶妱
丂崪奿嬝偱偼嬝彫朎懱乮SR乯偺Ca2+梀棧僠儍僱儖偱偁傞RyR1偑奐岥偟偰嬝廂弅偵昁梫側Ca2+傪梀棧偟傑偡丅RyR1偼SR枌忋偵2楍偵攝楍偟丄堦偮偍偒偵T娗枌偺僕僸僪儘僺儕僕儞庴梕懱乮DHPR乯偲憡屳嶌梡偟偰偄傑偡乮恾1A乯丅RyR1偼2偮偺儌乕僪偱奐岥偟傑偡丅DHPR偲憡屳嶌梡偟偨RyR1偑扙暘嬌偵傛傝奐岥偡傞扙暘嬌桿敪惈Ca2+梀棧乮DICR乯偲Ca2+偑捈愙寢崌偟偰奐岥偡傞Ca2+桿敪惈Ca2+梀棧乮CICR乯偱偡乮恾1B乯丅惗棟揑側嬝廂弅偱偼RyR1偼DICR偱奐岥偟傑偡偑丄偙偺嵺丄乽DHPR偲憡屳嶌梡偟偰偄側偄RyR1偑CICR偵傛偭偰奐岥偟偰Ca2+僔僌僫儖偺憹暆傪婲偙偡乿壜擻惈偑媍榑偝傟偰偄傑偟偨乮恾1A塃丄僋僄僗僠儑儞儅乕僋乯丅
丂偦偙偱丄RyR1偺Ca2+寢崌晹埵偵懚嵼偡傞僌儖僞儈儞巁乮E3896乯傪傾儔僯儞偵抲姺偟偨曄堎儅僂僗乮RyR1-E3896A乯傪嶌弌偟傑偟偨丅RyR1-E3896A儅僂僗偱偼DICR偼惓忢偵婲偙傝傑偡偑丄Ca2+偑寢崌偱偒側偄偨傔偵CICR偼梷惂偝傟偰偄傑偡乮恾1C乯丅偙偺儅僂僗偺Ca2+僩儔儞僕僄儞僩偍傛傃嬝廂弅偼栰惗宆儅僂僗偲摨摍偱偟偨丅偟偨偑偭偰丄CICR偑DICR偺憹暆婡峔偲偟偰婡擻偡傞偲偄偆壖愢偼斲掕偝傟傑偟偨乮恾1D嵍乯丅
丂RyR1偼埆惈崅擬徢乮Malignant Hyperthermia, MH乯傊偺娭梌偑帵嵈偝傟偰偄傑偡丅MH偼媧擖杻悓栻偵傛傝嬝嫮捈偲崅懱壏傪堷偒婲偙偡嬝幘姵偱丄偦偺庡側尨場偼RyR1堚揱巕偺曄堎偵傛傞僠儍僱儖妶惈偺堎忢槾恑偱偡丅RyR1-E3896A儅僂僗傪埆惈崅擬徢儌僨儖儅僂僗乮RyR1-R2509C乯偲岎攝偟偰摼偨巈儅僂僗偼MH偵懳偡傞掞峈惈傪帵偟傑偟偨丅偟偨偑偭偰丄CICR偺埆惈崅擬徢傊偺娭梌偑柧妋偵側傝傑偟偨乮恾1D塃乯丅
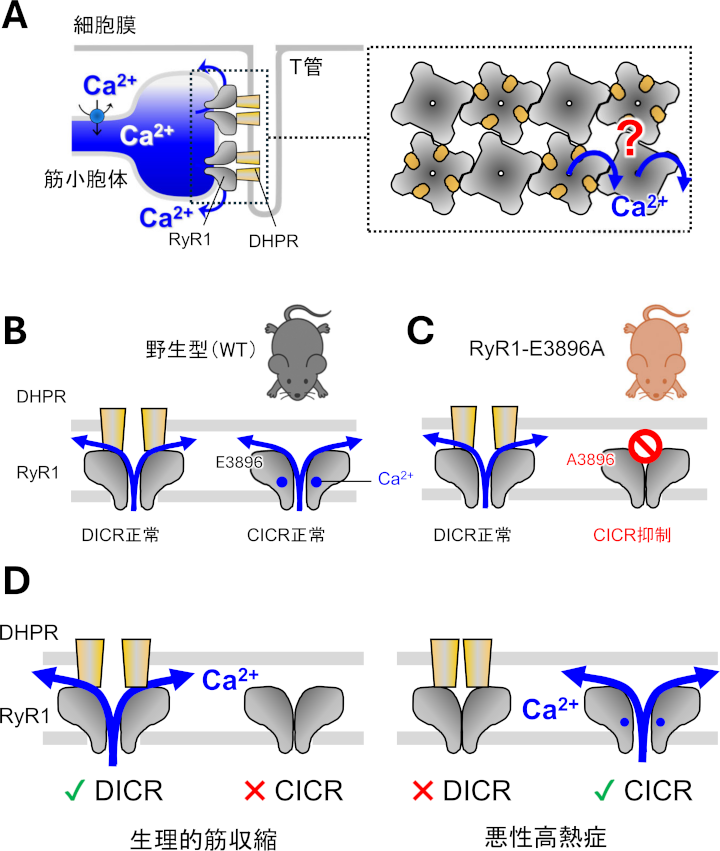 |
| 恾1. A. 崪奿嬝偵偍偗傞SR偐傜偺Ca2+梀棧婡峔丅RyR1偼SR枌忋偵2楍偵攝楍偟丄堦偮偍偒偵T娗枌偺DHPR偲憡屳嶌梡偡傞丅B. RyR1偼DICR偲CICR偺2庬椶偺奐岥儌乕僪傪桳偡傞丅C. RyR1-E3896A儅僂僗偱偼DICR偼惓忢偵婲偙傞偑丄CICR偼梷惂偝傟偰偄傞丅D. 杮尋媶偺惉壥丅CICR偼惗棟揑側嬝廂弅傊偺婑梌偑傎偲傫偳側偄偺偵懳偟偰丄埆惈崅擬徢偺敪徢偵娭梌偡傞丅 |
嶲峫暥專
- Kobayashi T, Yamazawa T, Kurebayashi N, Konishi M, Tanihata J, Sugihara M, Miki Y, Noguchi S, Inoue YU, Inoue T, Sakuraia T, Murayama T. RyR1-mediated Ca2+-induced Ca2+ release plays a negligible role in excitation-contraction coupling of normal skeletal muscle. Proc Nat Acad Sci. 122: e2500449122, 2025[Link]
B. 1宆儕傾僲僕儞庴梕懱偵懳偡傞慾奞栻扵嶕偲帯椕栻奐敪
丂埆惈崅擬徢乮MH乯偼RyR1僠儍僱儖妶惈偑槾恑偡傞偙偲偱敪徢偡傞偨傔丄RyR1慾奞栻偼帯椕栻偺桳椡側岓曗偲側傝傑偡丅尰嵼丄僟儞僩儘儗儞偑MH帯椕栻偲偟偰巊梡偝傟偰偄傑偡偑丄悈梟惈偑掅偔丄寣拞敿尭婜偑挿偄偲偄偆寚揰偑偁傝傑偡丅偟偨偑偭偰丄傛傝椙偄帯椕栻偑媮傔傜傟偰偄傑偡丅 丂巹偨偪偼曄堎宆RyR1傪HEK293嵶朎偵敪尰偝偣傞偲Ca2+儕乕僋傪婲偙偟偰彫朎懱撪Ca2+擹搙偑掅壓偡傞尰徾乮恾1E乯偐傜拝憐傪摼偰丄彫朎懱撪Ca2+擹搙應掕傪棙梡偟偨RyR1慾奞栻扵嶕僔僗僥儉傪奐敪偟傑偟偨丅婡擻婛抦壔崌暔儔僀僽儔儕偺僗僋儕乕僯儞僌偵傛傝丄僆僉僜儕儞巁傪娷傓4庬椶偺RyR1慾奞栻傪摨掕偟傑偟偨乮恾2乯丅
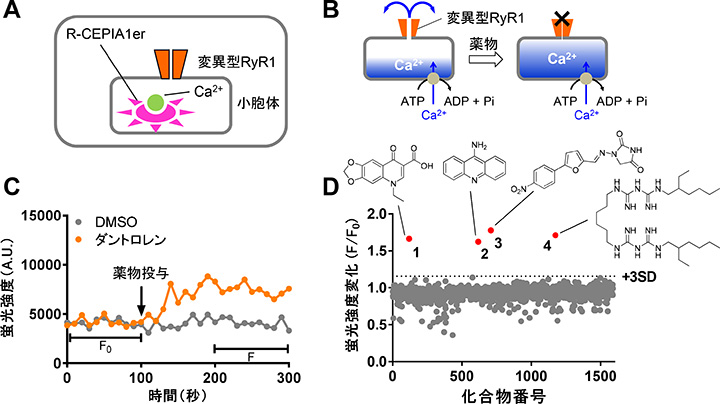 |
| 恾2. A. 僗僋儕乕僯儞僌偵梡偄偨嵶朎丅HEK293嵶朎偵曄堎宆RyR1偲彫朎懱撪Ca2+僀儞僕働乕僞偱偁傞R-CEPIA1er傪埨掕敪尰偟偨丅B. 僗僋儕乕僯儞僌偺僐儞僙僾僩丅曄堎宆RyR1傪敪尰偡傞偲Ca2+儕乕僋偵傛傝彫朎懱撪Ca2+擹搙偑掅壓偡傞丅RyR1慾奞栻偼Ca2+儕乕僋傪巭傔丄彫朎懱撪Ca2+擹搙偑忋徃偡傞丅C. 僐儞僩儘乕儖栻暔傪梡偄偨應掕椺丅RyR1慾奞栻偺僟儞僩儘儗儞偼彫朎懱撪Ca2+擹搙傪忋徃偝偣偨丅D.壔崌暔儔僀僽儔儕傪梡偄偨僗僋儕乕僯儞僌寢壥丅4庬椶偺壔崌暔偑僸僢僩偟偨丅1: 僆僉僜儕儞巁丄2: 傾儈僲傾僋儕僕儞丄3: 僟儞僩儘儗儞丄4: 傾儗僉僔僕儞丅 |
丂僆僉僜儕儞巁傪峔憿揥奐偟偨偲偙傠丄恊榓惈偑旕忢偵崅偄Compound 1乮Cpd1乯傪尒弌偡偙偲偵惉岟偟傑偟偨丅Cpd1偼埆惈崅擬徢儌僨儖儅僂僗偵懳偟偰桪傟偨帯椕岠壥傪帵偟傑偟偨乮恾3乯丅Cpd1偼僟儞僩儘儗儞偵斾傋偰惗棟怘墫悈傊偺梟夝搙偑崅偔丄寣拞敿尭婜偑抁偄偙偲偐傜僟儞僩儘儗儞偺寚揰傪崕暈偟偨怴偟偄埆惈崅擬徢帯椕栻偵側傞壜擻惈偑偁傝傑偡丅
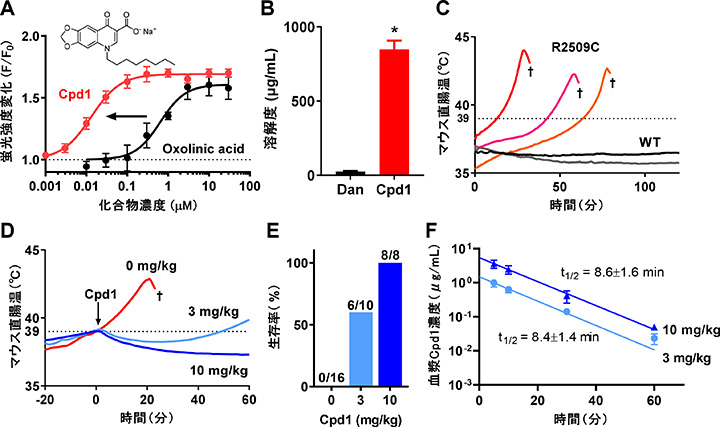 |
| 恾3. A. Cpd1乮僆僉僜儕儞巁桿摫懱乯偼僆僉僜儕儞巁偵斾傋偰RyR1慾奞妶惈偑70攞嬤偔忋徃偟偨丅B. Cpd1偼僟儞僩儘儗儞偵斾傋偰惗怘傊偺梟夝搙偑30攞埲忋崅偐偭偨丅C. 埆惈崅擬徢儌僨儖儅僂僗乮RyR1-R2509C乯偼僀僜僼儖儔儞杻悓偵傛傝懱壏偑忋徃偟偰巰朣偡傞丅D, E. Cpd1偼懱壏忋徃傪梷惂偟丄惗懚棪傪忋徃偟偨丅F. Cpd1偺寣拞敿尭婜偼10暘埲撪偲旕忢偵抁偐偭偨丅 |
嶲峫暥專
- Murayama, T, Kurebayashi N, Ikegami-Yuasa M, Mori S, Suzuki Y, Akima R, Ogawa H, Suzuki J, Kanemaru K, Oyamada H, Kiuchi Y, Iino M, Kagechika H, Sakurai T: Efficient high-throughput screening by ER Ca2+ measurement to identify inhibitors of ryanodine receptor Ca2+-release channels. Mol Pharmacol, 94: 722-730, 2018.[Link]
- Murayama T, Kurebayashi N. Assays for modulators of ryanodine receptor (RyR)/Ca2+ release channel activity for drug discovery for skeletal muscle and heart diseases. Curr Protoc Pharmacol, 87: e71, 2019.丂[Link]
- Mori S, Iinuma H, Manaka N, Ishigami-Yuasa M, Murayama T, Nishijima Y, Sakurai A, Arai R, Kurebayashi N, Sakurai T, and Kagechika H. Structural development of a type-1 ryanodine receptor (RyR1) Ca2+-release channel inhibitor guided by endoplasmic reticulum Ca2+ assay. Eur J Med Chem, 179: 837-848, 2019.[Link]
- Yamazawa T, Kobayashi T, Kurebayashi N, Konishi M, Noguchi S, Inoue T, Inoue YU, Nishino I, Mori S, Iinuma H, Manaka M, Kagechika H, Uryash A, Adams J, Lopez JR, Liu X, Diggle C, Allen PD, Kakizawa S, Ikeda K, Lin B, Ikemi Y, Nunomura K, Nakagawa S, Sakurai T, Murayama T. A novel RyR1-selective inhibitor prevents and rescues sudden death in mouse models of malignant hyperthermia and heat stroke. Nat Commun, 12: 4293, 2021[Link]
- Ishida R, Mori S, Murayama T, Nakamichi A, Chai X, Kurebayashi N, Iinuma H, Kagechika H. Development of a water-soluble ryanodine receptor 1 inhibitor. Bioorg Med Chem, 74, 117027. 儕儞僋丗https://doi.org/10.1016/j.bmc.2022.117027[Link]
C. 崪奿嬝扙暘嬌桿敪惈Ca2+梀棧乮DICR乯嵞尰宯偺峔抸
丂崪奿嬝偱偼RyR1偼墶峴彫娗乮T娗乯枌偺僕僸僪儘僺儕僕儞庴梕懱乮DHPR乯偺Cav1.1僒僽儐僯僢僩偲憡屳嶌梡偡傞偙偲偱丄嬝嵶朎枌偺扙暘嬌偵傛傝旕忢偵懍偄Ca2+梀棧傪婲偙偟傑偡乮扙暘嬌桿敪惈Ca2+梀棧丄DICR乯乮恾4A乯丅DICR偺峔憿婎斦偼Cav1.1偲RyR1偑懠偺DICR峔惉場巕乮兝1a丄Stac3丄JP2乯偲嫟偵宍惉偡傞DICR暋崌懱偱偡乮恾4B乯丅偙傟傜偺峔惉場巕偺堚揱巕曄堎偼愭揤惈儈僆僷僠乕傗埆惈崅擬徢摍偺擄帯惈偺嬝幘姵偺尨場偲側傝傑偡偑丄曄堎偵傛傞幘姵敪徢婡峔偼傛偔暘偐偭偰偍傜偢帯椕栻傕懚嵼偟傑偣傫丅巹偨偪偼丄旕嬝嵶朎偵DICR暋崌懱傪嵞峔惉偡傞偙偲偱岠棪揑側DICR嵞尰宯偺奐敪傪栚巜偟傑偟偨丅
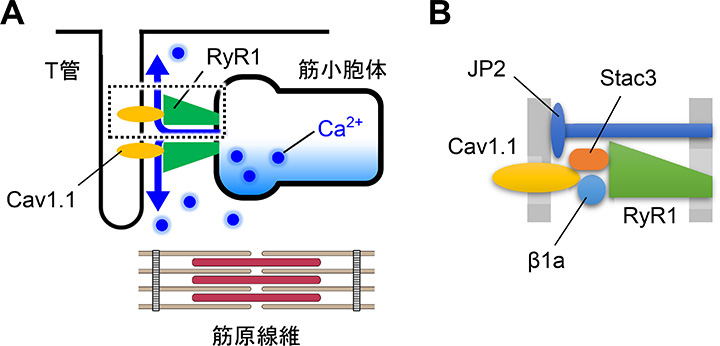 |
| 恾4. 崪奿嬝偺嫽暠廂弅楢娭婡峔偺柾幃恾丅A. T娗枌偺Cav1.1偺妶惈壔偑嬝彫朎懱枌偺RyR1僠儍僱儖傪奐岥偟偰廂弅偵昁梫側Ca2+偑梀棧偝傟傞丅B. DICR暋崌懱偺奼戝恾丅Cav1.1偲RyR1偼兝1a丄Stac3丄JP2摍偲DICR暋崌懱傪宍惉偟偰偄傞丅 |
丂RyR1偲寀岝彫朎懱撪Ca2+僀儞僕働乕僞偺R-CEPIA1er敪尰嵶朎傪梡偄偰丄DICR峔惉場巕傪夵曄僶僉儏儘僂僀儖僗儀僋僞乕偵傛傝堚揱巕摫擖傪峴偄傑偟偨丅偙傟偵傛傝丄傎偲傫偳慡偰偺嵶朎偵DICR峔惉場巕傪摫擖偡傞偙偲偵惉岟偟傑偟偨丅堚揱巕摫擖嵶朎偵懳偟偰崅僇儕僂儉梟塼偱巋寖偟偰DICR傪婲偙偡偲丄Ca2+梀棧偵傛傝彫朎懱撪Ca2+偑尭彮偟丄愒怓偺R-CEPIA1er寀岝偑掅壓偟傑偡乮恾5A,B乯丅峔惉場巕傪摫擖偟偨嵶朎偱偼僇儕僂儉擹搙偵埶懚偟偰R-CEPIA1er寀岝偑掅壓偟偨偺偵懳偟偰丄摫擖偟側偐偭偨嵶朎偱偼慡偔曄壔偟傑偣傫偱偟偨乮恾5C乯丅埲忋偺寢壥偐傜丄HEK293嵶朎偵偍偄偰DICR傪嵞尰偱偒偨偙偲偑暘偐傝傑偟偨丅
丂偙偺嵞尰宯傪梡偄偰幘姵曄堎偺昡壙傗栻暔僗僋儕乕僯儞僌偵棙梡偱偒傞偐偳偆偐傪丄婛曬偺幘姵曄堎懱偲栻暔傪梡偄偰挷傋傑偟偨丅愭揤惈儈僆僷僠乕偺尨場偲側傞Cav1.1曄堎懱乮P742Q乯偱偼僇儕僂儉擹搙埶懚惈偑戝偒偔掅壓偟偰丄DICR偑梷惂偝傟偰偄傑偟偨乮恾5D乯丅傑偨丄扨廂弅懀恑栻偺夁墫慺巁乮ClO4-乯偼DICR傪懀恑偟傑偟偨乮恾5E乯丅埲忋偺寢壥偼婛曬偲傛偔堦抳偟偰偍傝丄杮嵞尰宯偑幘姵曄堎懱偺昡壙傗栻暔僗僋儕乕僯儞僌偵棙梡偱偒傞偙偲傪帵偟偰偄傑偡丅
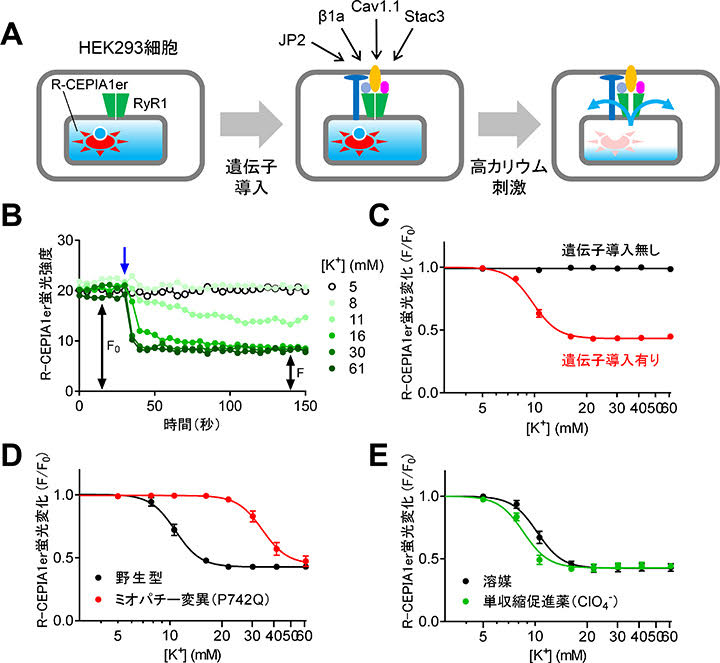 |
| 恾5. DICR嵞尰宯偺峔抸偲専徹丅A. 嵍丗RyR1偲R-CEPIA1er乮寀岝彫朎懱Ca2+僀儞僕働乕僞乯傪敪尰偡傞HEK293嵶朎偵DICR峔惉場巕乮Cav1.1丄兝1a丄Stac3丄JP2乯傪夵曄僶僉儏儘僂僀儖僗偱堚揱巕摫擖偟偨丅塃丗崅僇儕僂儉梟塼偵傛傝DICR偑婲偙傞偲彫朎懱撪Ca2+偑尭彮偟丄R-CEPIA1er寀岝偑掅壓偡傞丅B. DICR偺應掕椺丅崅僇儕僂儉梟塼偺揧壛乮栴報乯偵傛傝寀岝偑掅壓偟偨丅C. R-CEPIA1er寀岝偺僇儕僂儉擹搙埶懚惈丅堚揱巕摫擖偟偨嵶朎偺傒偱僇儕僂儉擹搙埶懚揑側寀岝偺掅壓偑尒傜傟偨丅D.儈僆僷僠乕曄堎乮Cav1.1-P742Q乯偺岠壥丅僇儕僂儉偺擹搙埶懚惈偑塃曽堏摦偟丄DICR偑梷惂偝傟偨丅E. 扨廂弅懀恑栻乮ClO4-乯偺岠壥丅DICR偑懀恑偝傟偨丅 |
嶲峫暥專
- Murayama T, Kurebayashi N, Numaga-Tomita T, Kobayashi T, Okazaki S, Yamashiro K, Nakada T, Mori S, Ishida R, Kagechika H, Yamada M, Sakurai T. A reconstituted depolarization-induced Ca2+ release platform for validation of skeletal muscle disease mutations and drug discovery. J Gen Physiol, in press..[Link]
D. 埆惈崅擬徢偍傛傃塣摦桿敪惈擬拞徢傪堷偒婲偙偡怴婯尨場堚揱巕偺扵嶕
埆惈崅擬徢傪堷偒婲偙偡尨場堚揱巕偲偟偰丄偙傟傑偱RYR1丄CACNA1S丄STAC3偑抦傜傟偰偄傑偟偨偑丄栺3妱偺姵幰偝傫偼堚揱巕夝愅傪偟偰傕忋弎偺堚揱巕偵婛抦曄堎偑傒偮偐傝傑偣傫丅忋婰偺3堚揱巕埲奜偵傕埆惈崅擬徢偵娭傢傞堚揱巕偑偁傞偲峫偊傜傟傑偡丅偦偙偱巹偨偪偼丄尨場堚揱巕偑摨掕偝傟偰偄側偄埆惈崅擬徢偺姵幰偝傫160柤偺DNA傪梡偄偰栐梾揑堚揱巕夝愅傪峴偄丄ASPH堚揱巕乮揮幨嶻暔: junctin乯曄堎偑埆惈崅擬徢偵娭傢傞偙偲傪偮偒偲傔傑偟偨丅
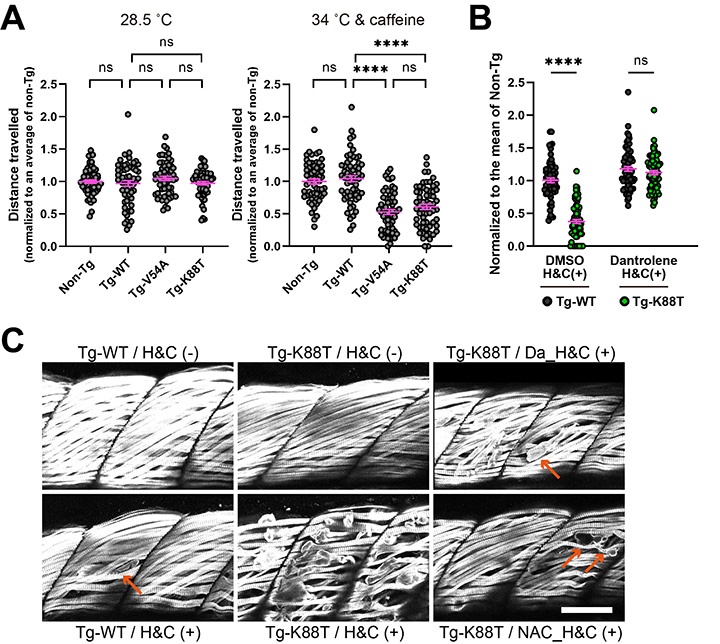 |
| 恾6. Junctin曄堎偑屄懱偵梌偊傞塭嬁偺専徹丅A. 僸僩偺栰惗宆junctin偲曄堎junctin傪夁忚敪尰偝偣偨僛僽儔僼傿僢僔儏傪嶌惉偟偨乮栰惗宆: Tg-WT, 曄堎宆: Tg-V54A, Tg-K88T乯丅捠忢偺帞堢壏搙乮28.5 亷乯偱偼Non-transgenic (Non-Tg)丄Tg-WT丄Tg-V54A丄Tg-K88T偵偍偄偰塣摦擻椡丒嬝椡偺嵎偼擣傔側偐偭偨丅僛僽儔僼傿僢僔儏傪僇僼僃僀儞擖傝34亷帞堢悈偱1帪娫帞堢偟偨屻丄梻擔偵塣摦擻椡傪應掕偟偨偲偙傠丄椉曄堎宆偼Non-Tg丄Tg-WT偵斾傋偰挊柧偵塣摦擻椡偺掅壓傪擣傔偨丅B. 埆惈崅擬徢帯椕栻偱偁傞RyR1慾奞嵻僟儞僩儘儗儞傪偁傜偐偠傔梊杊搳梌偟偨偲偙傠丄Tg-K88T僛僽儔僼傿僢僔儏傪僇僼僃僀儞擖傝34亷娐嫬乮H&C (+)乯偱帞堢偟偰傕梻擔偺塣摦擻椡偵掅壓傪擣傔側偐偭偨丅C. 僇僼僃僀儞擖傝34亷娐嫬乮H&C (+)乯偵敽偆嬝慻怐妛揑曄壔傪娤嶡偟偨丅Tg-K88T僛僽儔僼傿僢僔儏偱偼挊柧側嬝懝彎傪擣傔偨丅僟儞僩儘儗儞乮Da乯傕偟偔偼峈巁壔暔幙N-傾僙僠儖-L-僔僗僥僀儞乮NAC乯傪梊杊搳梌偟偨Tg-K88T僛僽儔僼傿僢僔儏偱偼丄嬝懝彎偺掱搙偑寉尭偟偨丅 |
嫽枴怺偄偙偲偵丄巹偨偪偑曬崘偟偨ASPH堚揱巕曄堎傪帩偮埆惈崅擬徢偺姵幰偝傫偼丄塣摦偵傛偭偰埆壔偡傞擔忢揑側嬝醶澒傗丄塣摦拞偺媫寖側懱壏忋徃偍傛傃堄幆忈奞偲偄偆丄塣摦桿敪惈擬拞徢偺徢忬傕暪敪偟偰偄傑偟偨丅偙偺偙偲偐傜丄ASPH堚揱巕堎忢偼塣摦桿敪惈擬拞徢偺尨場偵傕側傞偙偲丄埆惈崅擬徢偲塣摦桿敪惈擬拞徢偺儊僇僯僘儉偵嫟捠揰偑偁傞偙偲偑帵嵈偝傟傑偟偨丅
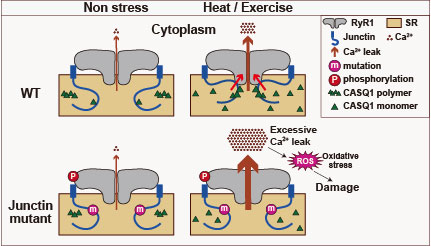 |
| 恾7. Junctin曄堎偵傛傞埆惈崅擬徢/塣摦桿敪惈擬拞徢偺儊僇僯僘儉偺採彞丅埨惷忬懺乮Non stress乯偱偺RyR1傪夘偟偨Ca2+儕乕僋偼栰惗宆丄曄堎宆偲傕偵彮検偱偁傞丅崅壏娐嫬壓偱塣摦偡傞偲乮Heat/Exercise乯丄RyR1傪夘偟偨Ca2+儕乕僋偑憹偊傞偑丄栰惗宆junctin偼calcequestrin(CASQ1)偲嫤摨偟偰RyR1偵摥偒偐偗丄Ca2+偑儕乕僋偟偡偓側偄傛偆偵挷惍偡傞丅曄堎宆junctin偼CASQ1偲楢実偱偒側偄偨傔RyR1傪惂屼偟偒傟偢丄憹壛偟偡偓偨Ca2+儕乕僋偑嬝懝彎傪堷偒婲偙偡偲峫偊傜傟傞丅 |
嶲峫暥專
- Endo Y, Groom L, Celik A, Kraeva N, Lee CS, Jung SY, Gardner L, Shaw MA, Hamilton SL, Hopkins PM, Dirksen RT, Riazi S, Dowling JJ. Variants in ASPH cause exertional heat illness and are associated with malignant hyperthermia susceptibility. Nat Commun, 13: 3403, 2022. PMID: 35697689. [Link]